

Pig model predicts the full therapeutic potential of exon skipping in Duchenne muscular dystrophy
18.07.2023
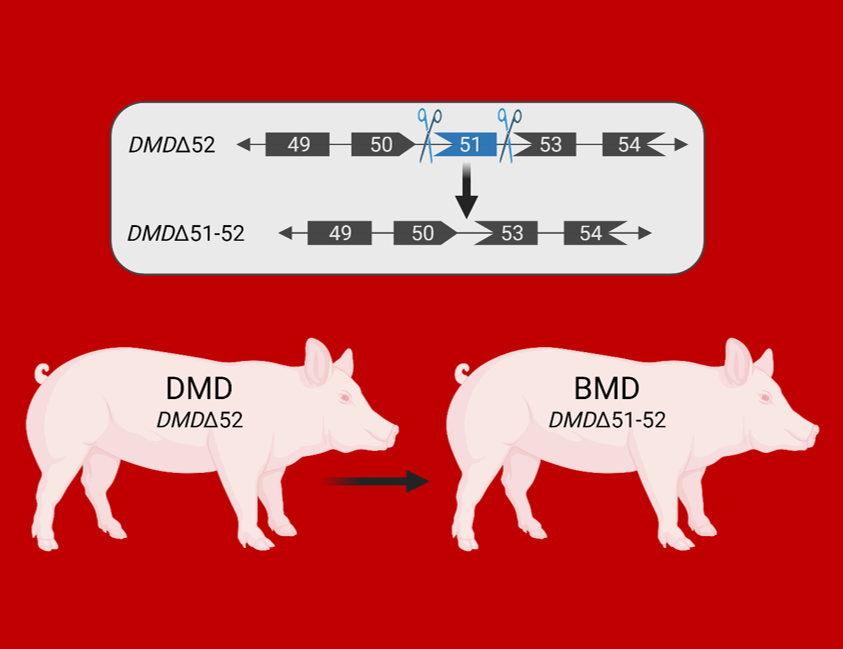
Duchenne muscular dystrophy (DMD) is a fatal X-linked disease caused by mutations in the DMD gene, leading to complete absence of dystrophin and progressive degeneration of skeletal musculature and myocardium. The majority of DMD mutations are deletions of one or more exons, with a hotspot in the range of exons 45 to 55. Frame-shift mutations lead to the formation of a non-functional, truncated dystrophin or the complete absence of dystrophin due to premature stop-codons and - as a consequence - to severe muscular dystrophy. A prominent example is the loss of exon 52, which resembles one of the most frequent DMD mutations in humans. Therefore, we developed a porcine DMD model with this mutation, which mimics the biochemical, clinical and pathological hallmarks of the human disease but develops them in an accelerated mode. In DMD patients and in the corresponding pig model with a deletion of DMD exon 52 (DMDΔ52), expression of an internally shortened dystrophin can be achieved by skipping of DMD exon 51 to reframe the transcript. However, current delivery strategies for antisense oligonucleotides or gene editing tools limit exon skipping to a proportion of (cardio-)myocytes; the full therapeutic potential remains thus unclear. Therefore, we generated a model of ubiquitous correction of DMD by systemic deletion of exon 51 in the dystrophic pig model lacking DMD exon 52. Molecular, pathological, and clinical alterations were largely rescued, supporting the optimization of exon skipping strategies as a clinically relevant goal. Due to the high susceptibility of pigs to dystrophic muscle lesions, DMDΔ51-52 pigs resembling a form of Becker muscular dystrophy (BMD) will uncover long-term outcomes within a reasonable time frame. This new study demonstrates that genetically tailored pig models are not only useful for studying disease mechanisms but also for predicting the best possible outcomes of targeted therapies.
https://doi.org/10.1073/pnas.2301250120